
More Information
Submitted: July 28, 2021 | Approved: August 26, 2021 | Published: August 27, 2021
How to cite this article: Nagy A, Sergi CM, de Nanassy J, Abbot LS, Arnoldo A, et al. A novel case of an infantile fibrosarcoma-like tumor with KIAA1549-BRAF translocation and an oncogenic NF2p.Q459* SNV with potential clinical significance. Arch Pathol Clin Res. 2021; 5: 016-019.
DOI: 10.29328/journal.apcr.1001023
Copyright License: © 2021 Nagy A, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

A novel case of an infantile fibrosarcoma-like tumor with KIAA1549-BRAF translocation and an oncogenic NF2p.Q459* SNV with potential clinical significance
Anita Nagy1, Consolato M Sergi3, Joseph de Nanassy3, Lesleigh S Abbot4, Anthony Arnoldo5, Cynthia Hawkins1,2,6 and Bo-Yee Ngan1,2*
1Department of Paediatric Laboratory Medicine, Division of Pathology, The Hospital for Sick, Children, Canada
2Department of Pathobiology and Laboratory Medicine, University of Toronto, Toronto, Ontario, Canada
3Anatomic Pathology Division, Children’s Hospital of Eastern Ontario, University of Ottawa, Ottawa, Ontario, Canada
4Department of Pediatrics, Division of Hematology/Oncology, Children’s Hospital of Eastern Ontario University of Ottawa, Ottawa, Ontario, Canada
5Department of Paediatric Laboratory Medicine, Laboratory of Molecular Diagnosis, Division of Pathology, The Hospital for Sick Children, Canada
6Director, Department of Paediatric Laboratory Medicine, Laboratory of Molecular Diagnosis, Division of Pathology, Canad
*Address for Correspondence: Bo-Yee Ngan, Division of Pathology, Department of Paediatric Laboratory Medicine, The Hospital for Sick Children, 555 University Avenue, Toronto, Ontario, Canada M5G 1X8. Email: [email protected]
We report a case of a right gluteal mass from the sacroiliac joint to the knee of an infant girl. Biopsy showed histopathological features similar to infantile fibrosarcoma (IFS). However, unlike most IFS, no ETV6-NTRK3 fusion gene abnormality was detected. Molecular analysis with TruSight RNA Pan-Cancer Panel detected the presence of KIAA1549-BRAF translocation and an oncogenic NF2p.Q459* SNV with potential clinical significance. A review revealed that the combination of this patient’s tumor site with the presence of a KIAA1549-BRAF translocation abnormality and an accompanying single nucleotide variant has not been previously described. The detection of this translocation abnormality raises the possibility that the spindle cell tumors in infants with an absence of the ETV6-NTRK3 fusion gene abnormality might have a distinct pathogenetic mechanism different from the previously known IFS and congenital mesoblastic nephroma. Furthermore, the discovery of BRAF translocation and its aberrant signaling of the mitogen-activated protein kinase (MAPK) pathway in this tumor contributes to the promise of clinical benefit of using the MEKi trametinib for the treatment of progressive disease that is refractory to conventional chemotherapy.
Infantile fibrosarcoma (IFS) is a rapidly growing and locally aggressive painless mass most commonly occurring in the extremities of young children under the age of two years. Most commonly ETV6-NTRK3 fusion is detected in IFSwhich it is therefore used as a diagnostic marker. Though number of new gene alterations have been described in recent years [1] and increases the genetic spectrum of IFS-like tumours IFS-like tumors represent a subset of childhood sarcomas with similar morphological features as IFS but without harboring the characteristic ETV6-NTRK3 fusion. These alternative rearrangements involve other kinase genes, such as NTRK3, NTRK2, NTRK1, BRAF and MET with having various fusion partners [1,2]. Currently, these tumorsare included with IFS category in 2020 WHO classification of soft tissue and bone tumours [2]. We report a case with IFS-like morphology with KIAA1549–BRAF fusion with an oncogenic NF2p.Q459* single nucleotide variant (SNV), which to our knowledge, is the first case of this kind reported in the limb.
Clinical history
A three-month old appropriately grown and developed female was prematurely born at 28 weeks’ gestation via emergency Caesarean section due to full length cord prolapse. Antenatal history included three episodes of antepartum hemorrhage and one-week history of premature rupture of the membranes prior to delivery.
At one month of age, the patient was noted to have developed an asymmetric swelling on the right buttock. On imaging (US, MRI) a large heterogeneous soft tissue mass was demonstrated, measuring 8.8. x 3.3. x 4.9 cm, with a focal cystic component up to 2.4 cm. The mass mainly involved the gluteus maximus and extended within the pelvis to the obturator internus and piriformis muscles and down to the posterior aspect of the right thigh to the knee level. The lesion was in close contact with the right-sided iliac vessels and their branches, the rectum and the urinary bladder. The right femur showed focal mild periosteal reaction only. There were no lung metastases identified on CT. The child was started on VAC chemotherapy, comprising vincristine, dactinomycin and cyclophosphamide as per protocol, following initial histological diagnosis of IFS. Upon reviewing the family history, it was revealed that the patient’s father, aunt and grandmother had “fatty tissue deposits” in their extremities, the nature of these lesions, however, had been unknown.
Histopathology findings
The biopsy demonstrated a moderately cellular tumour comprising monomorphic spindled cells arranged into intersecting fascicules (Figure 1a). The cells had uniform round to oval nuclei with fine nuclear chromatin and occasional nucleoli. Mitotic figures were easy to see (Figure 1b) and proliferation index with Ki67 was increased (Figure 1c). Very rare lesional cells expressed desmin (Figure 1d). Pan-cytokeratin (AE1/AE3), smooth muscle actin, myogenin, CD34, S100 and SOX10 were all negative (not shown). Nuclear expression of INI1/BAF47 was retained (not shown). Occasional nuclei were positive with p53, and there was no staining with BRAF and pan-TRK (not shown).
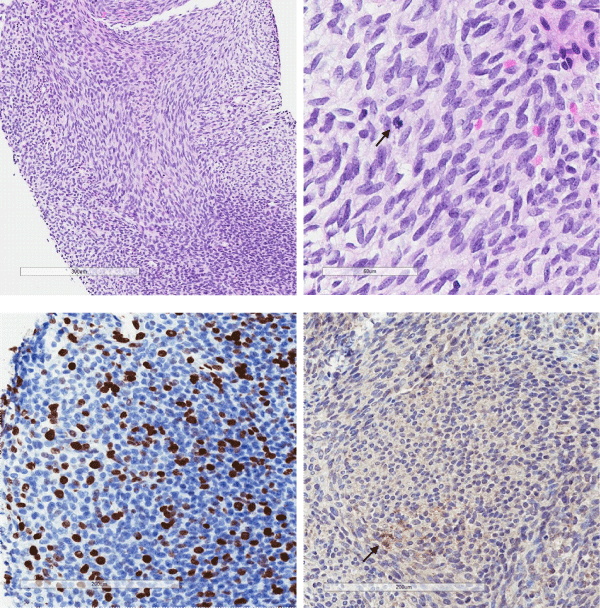
Figure 1: Moderately cellular tumour comprises spindle cells, arranged into intersecting fascicules (A, H&E, x20). The cells had uniform round to oval nuclei with fine nuclear chromatin and mitotic figures are easy to see (arrow) (B, H&E, x40). Immunostaining with Ki-67 showed increased proliferation activity (C, x20). Only a few lesional cells (arrow) were immunoreactive with desmin (D, x20).
Molecular findings
As initial genetic analysis for t(12;15) was negative, TruSight RNA Pan-Cancer Panel (1385 gene panel) was initiated. RNA was extracted from formalin-fixed paraffin-embedded tissue using RNAstorm FFPE RNA Isolation kit (Cell Data Sciences, Fremont, CA). Fragment length was assessed with an RNA 6000 chip on an Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA). RNA-sequencing library was prepared using 100 ng total RNA with the Illumina TruSight RNA Pan-Cancer Panel Kit (Illumina, Inc., San Diego, CA). The sample was subjected to targeted RNA sequencing on an Illumina MiSeq Dx platform.
Fusion calling
Reads were aligned to the hg19 human genomic scaffold using the Illumina STAR aligner (v2.6.1) or the DRAGEN aligner (v3.7.5). Fusion transcripts were identified using the Manta structural variant caller (v1.5.0) or the DRAGEN fusion caller (v3.7.5), respectively. Genes identified by both fusion callers were manually checked using Integrative Genomics Viewer (IGV) (v2.9.4) [3]. RNA fusion transcript was visualized using the St. Jude Cloud website [4]. Of note, a minimum of one gene in a putative gene fusion was needed to be present in the TruSight Pan-Cancer gene panel in order to be detected.
SNV calling
The Sequence was aligned to the hg19 human genomic scaffold using the Illumina STAR aligner (v2.6.1a) and single nucleotide variants were identified using the Strelka SNV / indel variant caller (v2.9.9). Genes identified as of potential diagnostic relevance were manually checked using IGV. Only “stop”, “truncation”, “frameshift”, “splice”, “deletion”, “insertion” and “missense” variant types were considered for analysis. Subsequently, only established oncogenic hotspot mutations found in COSMIC and/or ClinVar databases were reported. Variants of unknown significance were not reported. This RNA-based test could not distinguish between somatic and germline variants, and all variants were assumed to be somatic.
Using the above criteria, we detected KIAA1549 – BRAF fusion with the presence of an in-frame fusion between exon 15 of the KIAA1549 gene (NM_020910) and exon 11 of the BRAF gene (NM_004333). The kinase domain of BRAF, encoded by exons 11 to 18 was retained in the predicted fusion protein (Table 1, Figure 2, Figure 3). An oncogenic NF2p.Q459 SNV was also detected. The sequence change created a premature translational stop signal (p.Q459*) in the NF2 gene. It was predicted to result in an abnormal disruption/ termination of translation into protein product. We concluded this mutation to be a variant with potential clinical significance (Table 2). The missense variant of PMS2 was thought to be a variant of unknown clinical significance in this case (Table 2).
| Table 1: Fusion detection result by the automated fusion caller. | ||||||||
| Gene1 | Chr1 | Pos1 | Gene2 | Chr2 | Pos2 | Paired Read | Split Read | Score |
| Tier I: Fusions with Strong Clinical Significance | ||||||||
| KIAA1549 | chr7 | 138,552,721 | BRAF | chr7 | 140,481,493 | 1 | 5 | 0.596 |
| Table 2: Oncogenic hotspot SNVs identified by the automated caller. | |||||||||||||
| Gene | Chr | Pos | Depth | Ref | Alt | VAF | Variant type | Population databases | Genetic databases | Somatic databases | Predictive software | Base change | AA change |
| Tier II: Variants with Potential Clinical Significance | |||||||||||||
| NF2 | chr22 | 30,070,859 | 31 | C | T | 0.13 | nonsense | Not reported in gnomAD NR in dbSNP |
Not reported in ClinVar. | COSV58526762 | FATHMM prediction Pathogenic score 0.96) | r.1375c>u | p.Q459*a |
| Tier III: Variants of Unknown Clinical Significance | |||||||||||||
| PM2 | chr7 | 6,043,660 | 24 | G | A | 0.13 | missense variant | Not reporter in gnomAD NR in dbSNP |
Not reported in ClinVar. | COSV56220459 | FATHMM prediction Pathogenic score 0.9) | r.193c>a | p.L65I |
| a This sequence change creates a premature translational stop signal (p.Q459*) in the NF2 gene. It is predicted to result in an absent or disrupted protein product. | |||||||||||||
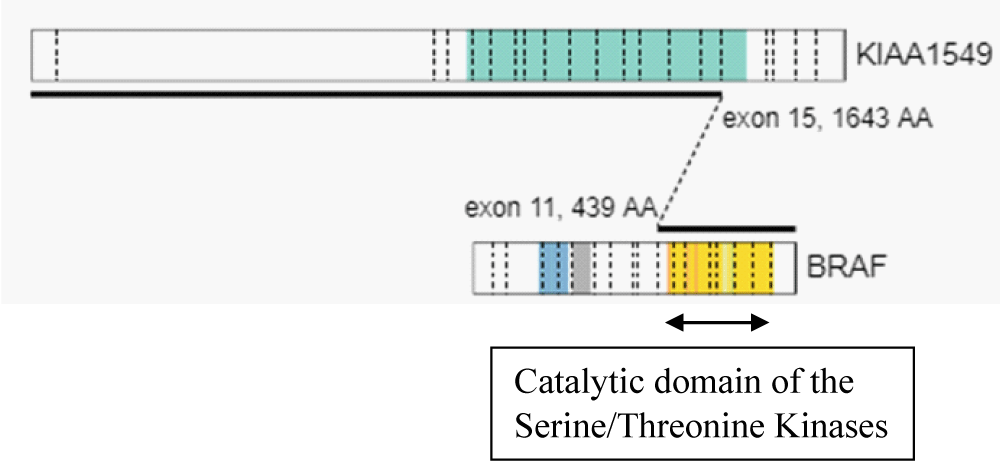
Figure 2: Fusion genomic breakpoints: in-frame fusion between exon 15 of the KIAA1549 gene (NM_020910) and exon 11 of the BRAF gene (NM_004333).

Figure 3: A KIAA1549/BRAF gene fusion was identified by targeted RNA sequencing
The most frequently detected gene fusion in IFS is ETV6-NTRK3, t(12:15)(p13;q25). Infantile spindle cell sarcomas with similar histopathological features that lack this fusion abnormality, developed through oncogenic activation of various other genes involved in kinase signalling pathway, have recently been reported (e.g.; NTRK3, NTRK2, NTRK1, BRAF, MET). Moreover, there is significant variation in gene fusion partners (e.g.; LMNA, TPR) [1,2], and breakpoint locations within the exons. Given the extent of molecular alterations associated with IFS, next-generation sequencing - based RNA and DNA analysis should be considered in fusion negative cases by FISH and RT-PCR.
One of these alternative molecular abnormalities involves the BRAF gene, which encodes a member of RAF family protein kinases, functioning downstream of RAS as part of the MAPK signaling pathway and facilitating cell proliferation, survival and transformation [5]. Kao, et al. reported 4 cases of pediatric spindle cell sarcomas with BRAF gene rearrangement in 2018. One patient presented with congenital mass in the thigh extending into the pelvis, two patients had pelvic mass and one patient had an extradural tumor with T6-T12 involvement. FISH detected these rearrangements and only the extradural case had further molecular testing confirming CUX1-BRAF fusion. Fusion partners in the remaining three cases remained unknown [6]. Rankin, et al. analysed 3633 pediatric tumour samples of extracranial solid tumours, brain tumours and hematological malignancies, of which 221 samples had BRAF alterations. In this study, KIAA1549-BRAF fusion was detected in three extracranial pediatric sarcoma cases, including cases of embryonal rhabdomyosarcoma, malignant peripheral nerve sheath tumor and sarcoma, NOS [5]. In a recent study of BRAF-altered spindle cell neoplasms with IFS-like morphology, Penning, et al. reported one case of KIAA1549-BRAF fusion in a three-month-old infant’s paraspinal mass [7].
Pediatric tumors with BRAF translocation are not limited to sarcomas in soft tissues. It has been known that congenital mesoblastic nephroma (CMN) and IFS share ETV6-NTRK3 gene fusion [8]. KIAA1549-BRAF gene fusion was also recently documented in a mixed CMN to analyze soft tissue tumours in infants [9]. These current findings of BRAF gene rearrangement in CMN and IFS, represent additional genetic overlap between the pathogenesis of these two neoplasms. Similarly, BRAF-translocation occurs in a number of low grade pediatric central nervous system tumors [10]. In pediatric low grade gliomas, translocation- related gain of function gene activation leads to an aberrant signalling of the MAPK. Early reports from clinical trials using small molecule inhibitors such as MEK inhibitors (MEKi) appears to be promising [11] in the treatment of pediatric low grade glioma. In light of finding of a similar pathogenetic mechanism in this sub-type of IFS in this report, consideration of a similar treatment strategy if this tumor is refractory to chemotherapy may become feasible.
In our case, there was an additional molecular event, involving NF2 gene, detected. NF2 encodes a tumor suppressor protein, Merlin (Moesin-ezrin-radixin-like protein). Loss of function mutations or deletions in NF2 cause neurofibromatosis type 2 (NF2), characterized by bilateral vestibular schwannomas. Additionally, patients can develop schwannomas of other cranial and peripheral nerves, as well as meningiomas and ependymomas. NF2 mutations also occur in spontaneous schwannomas and meningiomas, and other types of cancer, for example primary osteosarcoma, mesothelioma, glioma multiforme, hepato-biliary cancers, melanoma and medullary thyroid carcinoma [12]. There is very limited data available on the role of NF2 mutation or inactivation in cancer tumour genesis, though it might be relevant for prognosis and future chemotherapeutic approaches.
We report a right thigh mass in an infant with IFS-like morphology, KIAA1549-BRAF fusion and oncogenic NF2p.Q459* SNV. This BRAF fusion in soft tissue sarcomas is exceedingly rare, and our case is the second one reported in the literature. Considering alternative and less frequent gene arrangements in fusion–negative cases by FISH and initiation of further molecular analysis by TruSight RNA is critically important in pediatric soft tissue sarcomas as these patients might benefit from targeted therapy (e.g.; BRAF with MEKi). Further studies of IFS-like tumors with BRAF rearrangement are needed to characterize these lesions further and understand if these neoplasms represent a stand-alone pathologic entity with a different biologic behaviour or not. Additionally, NF2 mutation or inactivation in cancer tumour genesis needs more attention, as it might also be relevant for prognosis and management decisions.
- Davis JL, Antonescu CR, Bahrami A. Infantile fibrosarcoma. In WHO classification of tumours: Soft tissue and bone tumours. IARC. 2020; 3.
- Antonescu CR. Emerging soft tissue tumors with kinase fusions: An overview of the recent literature with an emphasis on diagnostic criteria. Genes Chromosomes Cancer. 2020; 59: 437–444. PubMed: https://pubmed.ncbi.nlm.nih.gov/32243019/
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, et al. Integrative genomics viewer. Nat Biotechnol. 2011; 29: 24–26. PubMed: https://pubmed.ncbi.nlm.nih.gov/21221095/
- McLeod C, Gout AM, Zhou X, Thrasher A, Rahbarinia D, et al. St. Jude Cloud: A Pediatric Cancer Genomic Data-Sharing Ecosystem. Cancer Discov. 2021; 11: 1082–1099. PubMed: https://pubmed.ncbi.nlm.nih.gov/33408242/
- Rankin A, Johnson A, Roos A, Kannan G, Knipstein J, et al. Targetable BRAF and RAF1 Alterations in Advanced Pediatric Cancers. The Oncologist. 2021; 26; e153-e163. PubMed: https://pubmed.ncbi.nlm.nih.gov/32918774/
- Kao YC, Fletcher CDM, Alaggio R, Wexler L, Zhang L, et al. Recurrent BRAF Gene Fusions in a Subset of Pediatric Spindle Cell Sarcomas: Expanding the Genetic Spectrum of Tumors With Overlapping Features With Infantile Fibrosarcoma. Am J Surg Pathol. 2018; 42: 28–38. PubMed: https://pubmed.ncbi.nlm.nih.gov/28877062/
- Penning AJ, Al-Ibraheemi A, Michal M, Larsen BT, Cho SJ, et al. Novel BRAF gene fusions and activating point mutations in spindle cell sarcomas with histologic overlap with infantile fibrosarcoma. Mod Pathol. 2021; 34: 1530–1540. PubMed: https://pubmed.ncbi.nlm.nih.gov/33850302/
- Knezevtch SR, Garnett MJ, Pysher TJ, Beckwith JB, Grundy PE, et al. ETV6-NTRK3 gene fusions and trisomy 11 establish a histogenetic link between mesoblastic nephroma and congenital fibrosarcoma. Cancer Res. 1998; 58: 5046-5048. PubMed: https://pubmed.ncbi.nlm.nih.gov/9823307/
- Wegert J, Vokuhl C, Collord G, Del Castillo Velasco-Herrera M, Farndon SJ, et al. Recurrent intragenic rearrangements of EGFR and BRAF in soft tissue tumors of infants. Nat Commun. 2018; 9: 2378. PubMed: https://pubmed.ncbi.nlm.nih.gov/29915264/
- Perreault S, Larouche V, Tabori U, Hawkin C, Lippé S, et al. A phase 2 study of trametinib for patients with pediatric glioma or plexiform neurofibroma with refractory tumor and activation of the MAPK/ERK pathway: TRAM-01. BMC Cancer. 2019; 19: 1250. PubMed: https://pubmed.ncbi.nlm.nih.gov/31881853/
- Selt F, van Tilburg CM, Bison B, Sievers P, Harting I, et al. Response to trametinib treatment in progressive pediatric low-grade glioma patients. J Neurooncol. 2020; 149: 499–510. PubMed: https://pubmed.ncbi.nlm.nih.gov/33026636/
- Petrilli AM, Fernández-Valle C. Role of Merlin/NF2 inactivation in tumor biology. Oncogene. 2016; 35: 537–548. PubMed: https://pubmed.ncbi.nlm.nih.gov/25893302/