
More Information
Submitted: 27 August 2020 | Approved: 16 September 2020 | Published: 17 September 2020
How to cite this article: Lee M, Tan GS, Tham CK, Lim KT. Detection of IDH mutations in cerebrospinal fluid: A discussion of liquid biopsy in neuropathology. Arch Pathol Clin Res. 2020; 4: 011-023.
DOI: 10.29328/journal.apcr.1001018
Copyright License: © 2020 Lee M, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Detection of IDH mutations in cerebrospinal fluid: A discussion of liquid biopsy in neuropathology
Ming Lee1*, Gek San Tan1, Chee Kian Tham2 and Kiat-Hon Tony Lim1
1Department of Anatomical Pathology, Singapore General Hospital, Singapore
2Department of Medical Oncology, National Cancer Centre, Singapore
*Address for Correspondence: Lee Ming, Department of Anatomical Pathology, Singapore General Hospital, 20 College Road, Academia, Level 7, Diagnostics Tower, 169856 Singapore, Tel: +65-65767560; Fax: +65 62276562; Email: [email protected]; [email protected]
Isocitrate dehydrogenase (IDH) mutations are a common event in secondary glioblastoma multiforme and lower-grade adult infiltrative astrocytomas and independently confer a better prognosis [1,2]. These are highly conserved mutations during glioma progression and thus also a useful diagnostic marker amenable to modern molecular sequencing methods. These mutations can even be detected in sites distant from the primary tumour. We use an illustrative case of a patient with radiologically suspected recurrent astrocytoma and negative histology, but positive IDH-mutated tumour DNA detected within CSF. Our results demonstrated the usefulness of liquid biopsy for recurrent glioma within the context of equivocal or negative histopathological results, whilst also showing the ability to detect a de-novo IDH-2 mutation not present in the previous resection. Building on this ‘proof-of-concept’ result, we also take the opportunity to briefly review the current literature describing the various liquid biopsy substrates available to diagnose infiltrative gliomas, namely the study of circulating tumour DNA, circulating tumour cells, and extracellular vesicles. We outline the current challenges and prospects of liquid biopsies in these tumours and suggest that more studies are required to overcome these challenges and harness the potential benefits of liquid biopsies in guiding our management of gliomas.
Adult infiltrative gliomas broadly comprise astrocytomas and oligodendrogliomas and are the most common malignant primary brain tumour [3]. Traditional grading and classification used to be carried out based on light microscopic features [3]. With marked advances in our ability to interrogate tumour tissue specimens utilising powerful next-generation gene sequencing methods [4,5], different molecular signatures can be distinguished [6], enabling differing treatment approaches and more accurate prognostication. Notable among these discoveries is the recognition of IDH mutations as an independent prognostic factor which is more common in low-grade astrocytomas, oligodendrogliomas and secondary glioblastomas. This mutation confers significantly improved survival in GBM patients [7-9]. Analysis of genetic markers in glioblastoma requires typically invasive surgery to obtain adequate amounts of tumour tissue to be useful. Moreover, glioblastomas are known to have intertumoral heterogeneity [10] and also undergo changes to their genomic profile in response to treatment [6,11]. Both these changes may be impractical to track with surgical tumour biopsies alone. To address these limitations, there has been avid interest in devising reliable and feasible methods of tracking and characterising important genetic markers of glioblastoma through blood and cerebrospinal fluid (CSF).
Herein, we present a case report of successful extraction and sequencing of IDH-1 mutation in ctDNA obtained from the CSF, and how it helped in establishing a diagnosis where histology failed. Beyond this, we also discuss the general utility and limitations of ctDNA analysis of CSF in glioma and briefly compare it with other methods of using circulating body fluids to elucidate the genome of malignant gliomas.
A 44-year-old woman presented initially with generalised tonic-clonic seizures. Neuroimaging revealed a large mass with cortical and subcortical T2/FLAIR hyperintensity in the left anteromedial temporal lobe, left uncus, hippocampus and Left frontal lobe. A resection was carried out. Microscopic examination showed an anaplastic astrocytoma, WHO Grade III. Immunohistochemistry showed it to be positive for IDH-1-R132H protein, the most common IDH-1mutation in adult gliomas. The patient was given temozolomide chemotherapy and cranial irradiation. She unfortunately developed wound infection, cerebrospinal fluid leak and recurrent encephalitis. Subsequent recurrence of intracranial tumour and development of drop metastases into the spinal cord was suspected on follow-up imaging. Open re-biopsy of the original tumour site showed only benign inflammatory changes. The suspicion of recurrent tumour remained high despite this, and cytologic examination of cerebrospinal fluid obtained via lumbar puncture was conducted. Initial cytopathological examination showed scant numbers of atypical, enlarged cells which were positive for GFAP. Next-generation sequencing (NGS) for ct DNA was carried out on the cerebrospinal fluid as well as the paraffin-embedded tissue from the prior cranial surgery. The methods and results of these are described in the next section. The NGS results confirmed that there remained circulating tumour DNA in the CSF, and by extrapolation, it was likely that there was sufficient tumour present, either remnant or recurrent, that had shed sufficient circulating tumour DNA containing the IDH mutation. A diagnosis of recurrent astrocytoma was able to be rendered based on these results. Before a second course of adjuvant therapy could be instituted, the patient deteriorated rapidly from infectious complications. She was treated palliatively and expired shortly after.
Immunohistochemistry
IDH1R132H mutation status for the pre-adjuvant, formalin-fixed, paraffin-embedded tumour tissue was confirmed via commercially available immunohistochemistry (Dianova, DIA-H09, dilution 1:200), with signal detection performed on the Bond Polymer Refine Detection Kit (Leica Biosystems, USA).
Next-generation sequencing
Next-generation sequencing was performed on crude cerebrospinal fluid (post-adjuvant) as well as the pre-adjuvant formalin-fixed, paraffin-embedded brain tumour tissue. Genomic DNA from both types of samples was extracted using the QIAamp DNA Mini kit and QIAamp DNA FFPE Tissue kit (QIAGEN), respectively. Library was constructed on 20 ng of each sample using a previously developed and validated custom panel (12) This panel contained primer pairs in three pools for hotspots and targeted regions of known genes associated with solid/epithelial cancers. Resulting amplicons were treated to partially digest, phosphorylate and ligate to Ion adaptors with barcoding, and purified. Quality and concentration of the libraries were determined using the Qubit 2.0 Fluorometer. Emulsion PCR and enrichment of template-positive Ion Sphere Particles (ISPs) was performed, followed by Amplicon sequencing on the Ion Torrent Personal Genome Machine (Ion PGM). Data is analyzed primarily using recommended in-built systems (Torrent Suite v5.2.2) and in-house bioinformatics pipelines v1.2.0 with reference genome hg19.
Panel design included Hotspots & targeted regions of the following 13 genes:
ALK, BRAF, CTNNB1, EGFR, ERBB2, ERBB4, IDH1, IDH2, KRAS, NRAS, PIK3CA, PTEN, and TP53.
Sequencing of the crude cerebrospinal fluid and pre-adjuvant FFPE brain tumour tissue were optimal with 98.58% coverage at minimum of 250x read depth for the gene panel; and up to 100% coverage at 1000x read depth for targeted hotspot exons of IDH1 (exons 4, 6) and IDH2 (exon 4) genes (Table 1).
| Table 1: Next-generation sequencing coverage and gene variant detection in the CSF and brain FFPE Tissue. | ||
| Specimen Type | Cerebrospinal Fluid (CSF) | Brain FFPE Tissue |
| Tumour Content | N.A. | 60% |
| Sequencing Coverage | IDH1: 100% (1000X) IDH2: 100% (1000X) Other genes: 98.58%(250X), 96.52%(500X), 80.92%(1000X) |
IDH1: 100% (1000X) IDH2: 100% (1000X) Other genes: 100.00%(250X), 100.00%(500X), 100.00%(1000X) |
| Gene variants detected | 1.IDH1 (NM_005896.3) Exon 4, c.395G>A; p.Arg132His Allelic Frequency: 44.1% 2.IDH2 (NM_002168.3) Exon 4, c.404C>T; p.Pro135Leu Allelic Frequency: 20.1% |
1.IDH1 (NM_005896.3) Exon 4, c.395G>A; p.Arg132His Allelic Frequency: 40.7% |
As seen in table 1, and graphically represented in figures 1a,b, an extremely high sequence coverage was achieved for IDH-1 and IDH-2 on next-generation sequencing conducted on both specimens, showing the variant allelic frequencies of both to be significantly high. This reaffirms the status of IDH mutations as a persistent, highly conserved mutation amenable to detection in biofluids after adjuvant therapy. Other than the presence of the canonical, more common IDH1 p.Arg132His (IDH-R132H) mutation in both samples, it was noteworthy that an additional new mutation in IDH2 (p.Pro135Leu) emerged in the post-adjuvant CSF sample. The significance of this latter finding is unknown as existing literature on IDH2 mutations in gliomas are confined to mutations at the amino acid site R172, which is analogous to R132 in IDH-1. [9,13]. Regardless of its current unknown significance, the detection of a new IDH2 mutation in the adjuvant specimen illustrates the importance and feasibility of liquid biopsies in monitoring the genomic landscape of gliomas over time.
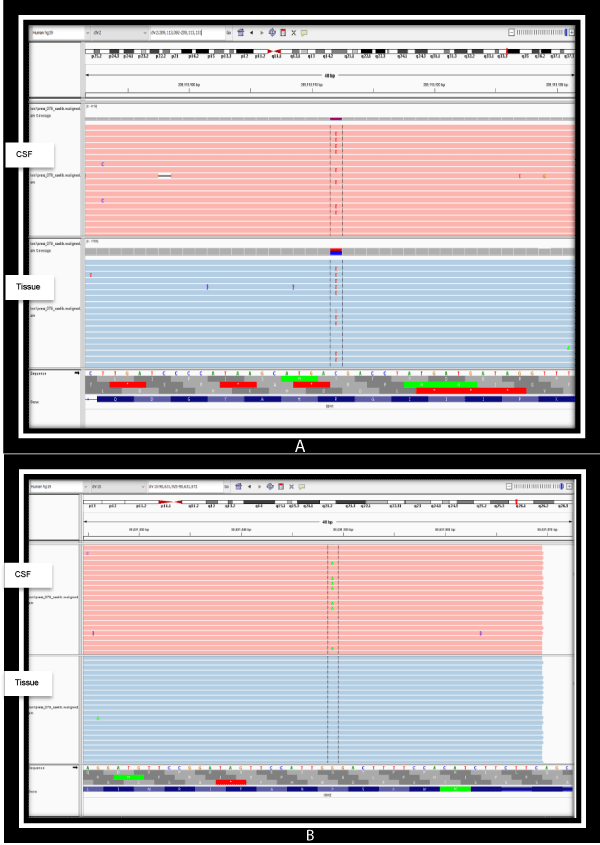
Figure 1: a) Detection of IDH1 (NM_005896.3) gene substitution variant (SNV), Exon 4, c.395G>A; p.Arg132His on both the CSF and brain FFPE tissue illustrated on the integrated genome viewer. b) Detection of IDH2 (NM_002168.3) gene substitution variant (SNV), Exon 4, c.404C>T; p.Pro135Leu on the CSF only illustrated on the integrated genome viewer.
Diagnosis of high-grade glioma presently requires histologic tissue sections, obtained via invasive surgery. However, diagnostic features may be elusive on histology due to extensive tumour necrosis, sampling error, prior chemoradiation therapy effects or masking by superimposed infection, as was the case with our patient. Longitudinal monitoring of such tumours is also crucial to understanding its response to adjuvant therapy effects and also to look for genetic events that enable tumour survival, facilitating the search for better treatment strategies to combat tumour survival [6].
Clinical response to treatment is conventionally monitored through neuroimaging. However, this is not as sensitive in discriminating pseudoprogression from tumour recurrence [14]. Moreover, it is impractical to constantly obtain brain tissue for genetic tumour profiling to study the genetic evolution of the tumour. Such genetic monitoring methods are better achieved through a ‘liquid biopsy’ of circulating body fluids. It has been known that malignant tumours shed, via various mechanisms, materials that contain DNA and RNA into systemic circulating fluids. Multiple methods exist for obtaining circulating tumour-derived genetic material. A schematic diagram of the various biomarkers amenable to study are illustrated briefly in figure 2, and discussed in further detail below.
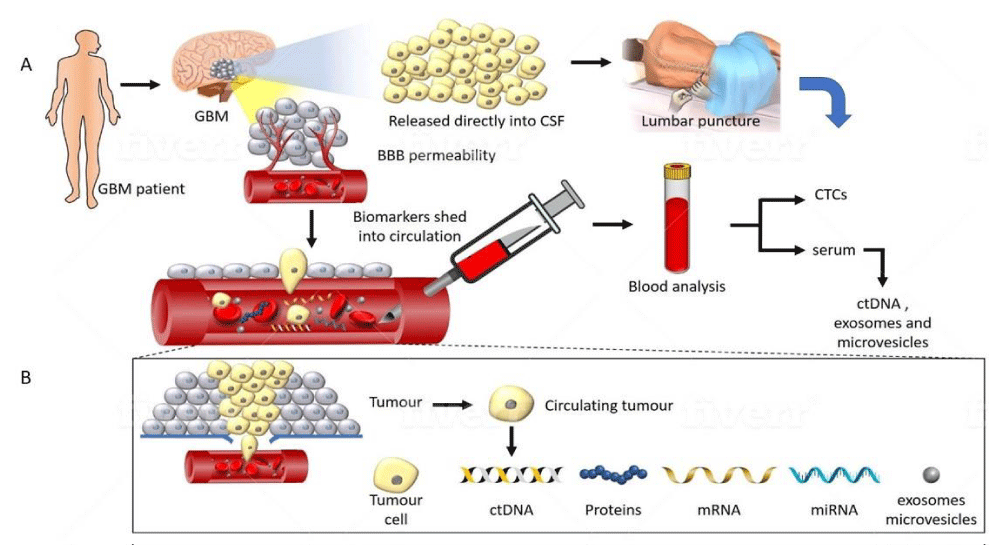
Figure 2: A representation of tumour components transported from a glioma into CSF and peripheral blood. (A) GBM disrupts the BBB allowing circulating biomarkers to enter peripheral blood. Alternatively, tumour proximity to a CSF-containing space obviates the need for passage through the BBB, allowing a potentially higher yield in CSF. (B) Detailed composition of the tumoural components detectable in the circulation. Several classes of molecules can be captured and studied, including CTC’s, extracellular vesicles, ct-DNA, mRNA and miRNA. These carry information such as mutational status, epigenetic changes such as methylation, and chromosomal abnormalities which can be sampled non-invasively.
Circulating tumour cells
CTC’s are tumour-derived cells that have entered the bloodstream (or in some special cases of malignant brain tumours, into the cerebrospinal fluid). They have the advantage of being representative of the genome of tumours known to be homogeneous, such as gastric carcinoma [15], and provide more analytic material for various ‘omics’ studies such as cell proteins and mRNA [16,17]. Quantification of CTC’s also is a reasonably accurate tool for gauging tumour burden, disease progression or regression [17]. However, the minuscule numbers of circulating tumour cells entering the systemic circulation make their study subject to sampling bias when characterising the heterogeneous genomic landscape within malignant tumours, particularly one as biologically aggressive as adult high-grade glioma [17-19].
The aforementioned problem of low CTC numbers [15,20,21] is particularly amplified in central nervous system tumours due to the blood-brain barrier and host immunity suppressing tumour migration and survival outside the neuroaxis. The short survival time of most glioma patients also contributes to the rarity of distant metastatic events [20]. However, cases of glioblastoma cells in the systemic circulation, and even extracranial metastases have been published [23-25]. This knowledge has guided newer strategies for purification, detection and subsequent analysis of circulating glioma cells, mostly exploiting the differing surface protein expression and biophysical properties of circulating tumour cells compared to circulating normal blood cells [16,22,26].
Although there is well-established clinical utility in using pure quantification methods of circulating tumour cells to prognosticate breast, renal, prostate and colon cancers, the evidence on applying such an approach to gliomas is lacking [17,20,22], reflecting the novelty of this approach. Studies that attempt to elucidate clinically useful genomic mutations from glioma CTC’s are also relatively lacking, with most studies of CTC’s in glioma focusing on their epithelial to mesenchymal transition abilities and their possible role in tumorigenesis [20,22,27]. Notably, there is a relatively new study by Krol and colleagues [28], using captured glioma CTC clusters to conduct exome sequencing, but purely as a method of confirming the presence of bona fide glioblastoma cells. The study did not mention using these CTC clusters to look for clinically useful molecular changes such as IDH mutation.
CTC’s in glioma patients have only very recently been studied in earnest, and the literature is still relatively sparse on the subject, making future, deeper study needed. The extant various published studies use different isolation and purification methods for CTC’s, making it difficult to compare results and to bridge the gap from experimental work to clinical utility at present. However, these studies show the potential of the application of CTCs as a liquid biopsy tool in GBM, and underscore a need for further investigation by larger trials.
Circulating tumour DNA
Both healthy and cancerous cells may release DNA into the circulation when they undergo apoptosis. Cell-free DNA (cfdna) consists of short lengths of DNA of 150 to 200 base pairs long, released directly into the extracellular compartment. Phagocytic mechanisms rapidly clear these fragments, and consequently they have a short half-life, reported to be 1.5 to 3 hours [22,29-31]. In cancer patients, a proportion of this cfDNA will contain circulating tumour DNA or ctDNA. The amount of ctDNA positively correlates with tumour burden and stage of disease [31]. Discriminating ctDNA from normal cfDNA is aided by the fact that the presence of somatic mutations defines tumour DNA. These somatic mutations, are unique to the genome of cancer cells when detectable, are useful in discriminating them from DNA derived from normal cells within the circulating fluids of an individual.
However, the detection technology has to be sufficiently sensitive for distinguishing mutant DNA from normal leucocyte-derived DNA, and also accurate enough for calculating the tumour mutation burden. Furthermore, ctDNA fragments have a short half-life as previously mentioned and require rapid sample processing. Provided detection methods are robust enough; ctDNA can detect mutations exclusive to the tumour. Theoretically, conventional sequencing methods such as Sanger sequencing and pyrosequencing could identify mutations in ctDNA if tumour DNA fragments in the sample were concentrated enough, which is only true in patients with later-stage disease where the tumour burden is high. Unfortunately, this means that technical challenges arise because for ctDNA to be useful, it has to be detected at low concentrations that typically represent only a small fraction (less than 1.0%) of total cfDNA [32]. These sensitivity limitations on detection of ctDNA in cancer patients have been surmounted due to the advancement and increased usage of digital genomic technologies, as well as the optimisation of next-generation sequencing (NGS) technologies. These technological advancements allow for extremely sensitive enumeration of a wide array of mutant DNA sequences in body fluids [33]. Before the introduction of techniques like digital polymerase chain reaction (PCR) [34] beads, emulsion, amplification, and magnetics (BEAMing) [34]. detection of cfDNA derived from tumours was inconsistently detected [33], with some authors suggesting that ctDNA was inferior to other circulating biomarkers [32,35,36].
The low detection sensitivity of brain tumour derived-ctDNA in plasma has been attributed to the presence of the blood-brain barrier. Although improvement in sequencing technologies has partially overcome the challenge of low ctDNA concentrations in plasma, sensitivity overall remains low. An earlier study in 2014 by Bettegowda, et al. detected IDH1, EGFR, TP53 and PTEN mutations in a limited subset (10%) of patients with glioblastoma [31]. More recent studies performing comprehensive ctDNA analysis on plasma utilising a highly sensitive and specific NGS assay yielded a better detection rate, approximately 50% in patients with advanced glioblastoma and showed that the ctDNA detection rate in gliomas may vary by grade and histopathology [29]. A similar NGS analysis of plasma-derived ctDNA from 171 patients (including 33 patients with glioblastoma), successfully determined the most frequent mutations in the TP53, EGFR, MET, PIK3CA and NOTCH1 genes [37]. In other studies, the most frequent somatic mutations detected using NGS platforms were those of TP53, NF1, EGFR1, MET, APC and PDGFRA genes together with amplifications of ERBB2, MET and EGFR, among others [38]. These results mirror the mutational spectrum elucidated from tissue samples of glioblastomas described in The Cancer Genome Atlas [39].
In addition to genomic changes, epigenetic changes are also detectable in glioblastoma blood samples. The most prognostically and predictively significant epigenetic change in glioblastoma in extensive use clinically to date is methylation in promoter MGMT gene, which confers increased tumour sensitivity to alkylating chemotherapy. This was observed in matched tissue and serum ctDNA from glioblastoma patients as well as correlated with clinical response to therapy [40]. Specifically, patients with increased serum levels of MGMT promoter methylation showed better clinical response and longer time to progression improved response and time to progression after treatment with alkylating agents. Recently, a sensitive assay based on immunoprecipitation has been reported as being able to study the methylome of small quantities of ctDNA and look for detect tumour-specific patterns early in the progression of the disease [40].
Other publications have tried to address the challenge of the blood-brain barrier by directly sampling cerebrospinal fluid. Wang, et al. analysed 35 CNS malignancies and found that all medulloblastomas, ependymomas and gliomas that abutted a CSF space yielded detectable circulating tumour DNA with mutations matching that obtained from the tissue sample (100% of 21 cases; 95% CI = 88% – 100%), whereas no CSF-tDNA was detected in patients whose tumours were not directly adjacent to a CSF reservoir (p < 0.0001, Fisher’s exact test) [30]. When Wang, et al. [41] analysed the serum and CSF of patients with different grades of glioma, they detected methylation of the MGMT promoter region on methylation-specific PCR, in 38 patients with GBM out of 89 glioma patients (42.6%). As expected, reported sensitivity was higher in the CSF compared with blood samples (serum), with methylation detected in 19 out of 89 patients (21.3%) in serum samples, and in 26 out of 78 patients (33.3%) by using CSF. Of note, these values increased to 72% and 41% for CSF and serum respectively in patients with postoperative residual tumours, possibly due to the disruption of tumour and blood-brain barrier during surgery.
De Mattos-Arruda and collaborators found mutations in IDH1/2, EGFR, PTEN, FGFR2 and ERBB2 genes in CSF, and showed that CSF ctDNA landscape is modulated over time in response to treatments after the observation of dynamic changes in CSF ctDNA, which closely follows the treatment courses of patients with glioblastoma [41]. Thus, ctDNA from CSF could be used to find somatic mutations and monitor tumour burden. Bolstering these results, Miller and colleagues studied CSF from 85 glioblastoma patients via next-generation sequencing and found ctDNA was detected in 49.4% of patients and associated with disease burden and adverse outcome [42]. Moreover, the tumour genomic landscape represented by the CSF ctDNA included many alterations which closely mirrored those seen in the paired tissue biopsies.These included mutations in TP53 and IDH1/2, deletions of CDKN2A and CDKN2B, and amplification of EGFR. When ctDNA mutations in IDH1/2 were found, the same mutation was found in the paired tumour. Additional analysis of CSF ctDNA revealed a broad spectrum of promoter mutations, copy number alterations and structural rearrangements. Important, clinically relevant alterations that occur in early tumorigenesis, such as 1p/19q codeletion, and mutations in the isocitrate dehydrogenase gene, were shared in all matched ctDNA/tumour pairs. Conversely, ctDNA of genes related to the growth factor receptor signalling pathways showed significant divergence with the initial tumour biopsy with time, highlighting the importance of ctDNA as a method of studying temporal variation in tumour genomic profiles.
The abovementioned results suggest that CSF may be better at capturing genomic alterations and is more sensitive method of detecting longitudinal glioblastoma genetic alterations than plasma ctDNA. Of note, promoter hypermethylation in MGMT, p16INK4a, TIMP-3 and THBS1 was detected at high frequencies in CSF, serum and tumour tissue, in all glioblastoma patients but not in any of the healthy individuals [40]. Hypermethylation in CSF and serum DNA was accompanied with methylation in the corresponding tumour tissues with 100% specificity [40]. These changes are interesting and may point a path toward liquid biopsies as a method of cancer screening.
Extracellular vesicles or exosomes: Extracellular vesicles (EVs) are small vesicles which are either produced in the cytoplasm with an intact endosomal membrane before subsequently fusing with the plasma membrane prior to extrusion from the cell, (exosomes); or alternatively, can be produced directly from the extracellular membrane via budding off (microvesicles). They can be taken up by other cells as well, thus providing signalling between various cell types, and are thought to play an important role in the development of therapy resistance in cancer [43]. EVs are present in nearly all bodily fluids and, importantly, represent a microcosm of the cytoplasmic contents from their host cancer cell, including proteins, membrane lipids, cell metabolites, micro-RNA and DNA [20,43]. The advantage of studying EV’s lies in the fact that the small vesicles have a protective membrane that shields their cargo from rapid destruction by circulating immune cells and enzymes, enabling enough time for their extraction and subsequent study. Some of the contents of interest in cancer are oncoproteins, oncogenic DNA mutation sequences and microRNA sequences.
Skog, et al. [44] have shown that EV’s are present and can be isolated from the serum of glioma patients, and that specific genetic alterations in the EGFR gene can be detected from serum-derived EV’s. Following the comparatively simpler task of detecting EGFRvIII mutations which are typically large deletions involving a large gene, further work in the EV field has been successful in demonstrating various other exosomal tumour-related molecules such as podoplanin [45], and also genes or gene products involving smaller, even single-nucleotide mutations—such as mutant IDH1 protein [34].
Other authors have also demonstrated the viability of using CSF samples to obtain EV’s. Chen, et al. [34], using BEAMing (beads, emulsion, amplification, magnetics) and droplet digital PCR analysis, have successfully demonstrated pathogenic mutated IDH1 mRNA transcripts within CSF of patients, with a sensitivity of 63% and a specificity of 100%. Similar sensitivity and specificity numbers have been published by others on detecting EGFRvIII mutant transcripts in CSF-derived EV’s [44].
In addition to directly transcribed RNA, other post-transcriptional events have been intensely studied in the field of EV’s, through the analysis of miRNAs. miRNA’s are short, 18 to 22-nucleotide-long sequences of RNA with essential roles in post-transcriptional regulation of gene expression. Coupled with the known function of EV’s in intercellular signalling, the role of miRNA’s in cancer diagnosis and prognosis is abundantly clear. A whole myriad of miRNA exists [46,47], and miRNA profiles are reasonably unique to the tissue of origin. These characteristics make miRNA an extremely promising class of biomarkers.
In tissue biopsies of glial tumours, miRNA profiles have been described, and various prognostic and predictive signatures have been reported. The proposed mechanism of the prognostic behaviour is via control of various post-transcriptional events important in the tumour’s biologic behaviour [10,46-48]. A number of miRNA have been shown to correlate with GBM survival, with one standing out in particular-- miR-21. Shi, et al. showed that the expression of miR-21 in GBM patients had a negative correlation with survival [49]. The high specificity and sensitivity of miRNA 21 were affirmed in a meta-analysis by Qu and colleagues, which demonstrated that miR-21 as a single miRNA exhibited high sensitivity and specificity [50].
Given that the correlations of miRNAs with therapy efficacy and tumour subtypes have already been established, these molecules could be targets for therapies, making the ability to detect them reliably an important area of research. As an example, miR-181 is directly involved in the downregulation of MGMT expression, and the level of miR-181 in the serum correlates with patients’ response to temozolomide [51].
Given that current efforts to modulate MGMT via administering pseudosubstrates to the MGMT enzyme have thus far shown little promise [52], the alternative strategy of upregulating miR-181 could decrease MGMT expression and thereby improve treatment response. This means that the detection and ongoing monitoring of miR-181could become increasingly important.
Why should we be considering liquid biopsy in surgical neuropathology practice?
Safety: Performing a lumbar puncture in patients with a brain tumour is usually safe and is done routinely for certain brain tumours as part of the staging criteria (for example, CNS lymphoma, medulloblastoma, germ cell tumours) [53]. In patients who have an inoperable tumour, a lumbar puncture offers an opportunity to genetically interrogate material shed by the tumour into a reservoir of fluid that can hold a significantly higher concentration of tumour DNA and is less prone to clearance by humoral immune system mechanisms in the plasma. In recurrences, a lumbar puncture is a simpler and safer procedure than a second craniotomy.
Capturing Tumour Heterogeneity: For our directed approach aimed at detecting a ubiquitous mutation such as IDH-1, which is known to occur early in gliomagenesis and already proven present in a prior tissue biopsy, consideration of a test’s ability to capture the heterogeneity of tumour tissue is not so important as the sensitivity of the method in detecting the target of investigation.
However, when considering the broader application of liquid biopsies in capturing rare mutation events, monitoring tumour biology and looking for mechanisms of tumour resistance, capturing the heterogeneous genomic makeup of the tumour becomes important.
The major limitation of tissue biopsy is representativeness, especially so in many neurosurgical specimens which may only be a small stereotactic biopsy of a large tumour. Glioblastomas are heterogeneous, with different areas of the same tumour showing different genetic profiles (i.e., intra-tumoural heterogeneity) [18].
Ideally, liquid biopsies should be able to capture this heterogeneity and enable a clinician to adapt their treatment strategies. However, different analytes differ in their ability to represent this heterogeneity. At present, it is unclear to what extent circulating tumour cells represent the main tumour, and their relevance for ongoing treatment decisions on the main tumour mass is uncertain [15,20].
In contrast, EV’s and circulating tumour DNA are thought to be produced universally throughout all cells, and by extension, all cancer cells. As such, these seem to provide better insight into tumour heterogeneity [15]
Tumour burden: Over the years, the discovery of circulating biomarkers has altered management of several malignancies, in some cases revolutionised them. Notable examples are cancer antigen (CA) 19-9 for pancreatic adenocarcinoma, carcinoembryonic antigen for colorectal carcinoma, and CA-125 for ovarian adenocarcinoma. The discovery and subsequent standardisation of these protein biomarkers have enabled reliable, clinically validated assessment of therapeutic response, disease burden, and a measure of biologic aggressiveness by their rate of change. Unfortunately, gliomas do not have a reliable peripherally detected protein biomarker, again due to the blood-brain barrier and the relative rarity of metastases outside the neuroaxis. Even if one was discovered, while it would undoubtedly be useful, it would be limited by a common feature of protein biomarkers—their persistence in circulation for weeks, as well as their elevation in the circulation weeks to months prior to relapse. While these would be a useful feature if the intention was purely to monitor long-term response and surveil for relapses, this persistence in the circulation limits a protein marker’s utility in assessing the accurate, day-to-day biologic response of the tumour to therapy and precludes precise, timely adjustments to therapy [54,55]. There are advantages to using ctDNA as a monitoring method instead of protein biomarkers or even imaging studies. For one, ctDNA has a short half-life (approximately 2 hours), allowing for evaluation of tumour changes in hours rather than the weeks to months that it takes before protein biomarker responses to therapy become detectable. A study by Diehl, et al. found that changes in ctDNA in colorectal carcinoma patients can predate those seen in imaging studies or using protein biomarkers by weeks to months [32].
Several investigational studies have shown that ctDNA can be a surrogate for tumour burden and that much like viral load changes (e.g. in viral hepatitis and HIV infections), levels of ctDNA correspond with clinical course. Studies in various solid extracranial malignancies (breast, melanoma, colon) have demonstrated the ability of this approach to more precisely characterise tumour dynamics in response to therapy [32,56,57]. These studies found that ctDNA levels increase rapidly in tandem with disease progression and also decline correspondingly after successful treatment with pharmacologic therapy or tumour resection. More relevant to the issue of primary intracranial neoplasms, a similar change in ctDNA is observed in glial tumours, as demonstrated by Mattos Arruda, et al. who have shown CSF ctDNA levels fluctuate in time and follow changes in brain tumour burden [41], providing a potential method of using circulating biomarkers to monitor brain malignancies. Further studies will be needed to validate this methodology for monitoring glioma tumour burden longitudinally.
Detecting residual disease: As of now, differentiating patients who are cured by resection, and those who have residual disease depends mainly on clinical and pathologic parameters, the most important being the TNM staging system. This system stratifies patients by risk for recurrence and possible benefit from adjuvant chemotherapy. However, this method uses parameters such as size, level of invasion, lymph node status and so on. Again, these are subject to, among other things, vagaries of sampling, pathologic interpretation and the resolution limit of current imaging technology. As such, these are probabilistic measures that attempt to quantify the potential risk of recurrence. There is, therefore, a risk of either subsequent under-or overtreatment of patients. With the emergence of liquid biopsy, its potential as an accurate measuring method of residual disease has been explored.
The best example to date has been in the cohort of 18 patients with colorectal cancer undergoing resection with curative intent [32]. The mutational profile was determined from each patient’s resected tumour, and this personal and unique molecular signature was then used to create a set of mutation-specific probes for each patient, before using these probes to detect and quantify ctDNA after surgery in these patients undergoing resection with curative intent. Over a follow-up period of 2 to 5 years, this study showed that ctDNA was shown to be sufficiently sensitive to detect minimal residual disease after surgical resection. All patients with detectable postoperative levels of ctDNA experienced recurrence, whereas all patients with undetectable postoperative levels of ctDNA remained disease-free. Similar studies have been performed examining RAS/RAF mutations in patients with resected colorectal cancer and shown a strong correlation between postoperative detection of mutant DNA in circulation and recurrence [58].
However, caution should be exercised when attempting to apply such a detection strategy to neuroaxis tumours, particularly infiltrative gliomas. An effort in using circulating DNA as a means of detecting and quantifying minimal residual disease for treatment triaging purposes is fraught with difficulties. The unique difficulty in attaining full resection with clear margins means that resection is rarely carried out with curative intent, as well as its confinement to the neuroaxis behind a relatively impermeable blood-brain barrier is reflected in its lack of TNM staging parameters.
As such, studies assessing ctDNA as a measure of glioma tumour burden [41] have not been able to extrapolate this to an assessment of ‘minimal residual disease’. However, a limited subset of tumours amenable to gross or near-total resection such as ependymoma [59] may benefit from the liquid biopsy revolution in this way. Future studies should be done to evaluate postoperative levels of ctDNA in ependymomas, providing a better way of monitoring recurrence and potentially avoiding unnecessary adjuvant therapy.
Monitoring of tumour resistance and heterogeneity: Development of resistance to chemotherapy results from the acquisition of mutations in genes. Defining these mechanisms of resistance to a targeted agent is often done using laboratory-cultured cell lines or xenografted laboratory animals, due to the impracticability of obtaining repeated biopsies in human subjects. In principle, every patient enrolled in a therapeutic clinical trial should undergo a tissue biopsy before initiation of the experimental therapy and after progression. Modern technology such as NGS has enabled us to search large expanses of the tumour genome to find genetic differences between the tissue collected before and after therapy. This will offer a snapshot of the predominant resistant clone of a portion of the lesion under examination. Recent studies have shown that liquid biopsies can be used effectively to monitor the emergence of multiple resistance clones during the course of treatment [20]. The genetic bases of secondary resistance to various targeted drugs have been elucidated in great detail [60-62]. The most extensive analyses are those involving Philadelphia chromosome–positive chronic myeloid leukaemia and gastrointestinal stromal tumours, both of which are treated with the tyrosine kinase inhibitor imatinib. Emergence of ABL kinase domain mutations have been extensively implicated in the pathogenesis of acquired resistance to imatinib, while in GIST, the best-accepted mechanism of resistance is the polyclonal expansion of multiple subpopulations harbouring different secondary KIT mutations [63].
Obtaining plasma from patients undergoing chemotherapy is trivially safe and straightforward with little contraindication, enabling unbiased study of the tumour genome post-therapy. This method has been applied to study ctDNA for the acquisition of secondary resistance. In a study by Murtaza, et al. [64] it was shown that genetic markers of resistance can be noninvasively tracked throughout the course of treatment in breast, ovarian, and lung cancers. Due to the short half-life of ctDNA, analysis of ctDNA in plasma samples obtained before and after treatment can ultimately provide insight into the molecular genetic changes of a patient’s tumour. This genetic picture includes quantitative information on dynamic changes in the mutation profile post-therapy as well as the heterogeneity resulting from therapeutic selective pressure. This understanding of the mechanisms of acquired resistance to drugs at the molecular level can be used to tweak the therapeutic regimen to include drugs that will suppress the tumour subpopulation responsible for the emergence of resistance. This success in tracking resistance at the molecular level via plasma for certain cancers opens new possibilities for studying resistance to chemotherapy in glial tumours via sampling of CSF fluid. Understandably of course, CSF is not as trivially obtainable as plasma, making it more difficult to decide how often, and for how long CSF can be studied this way. At present, there is no widely accepted genetic marker of tumour resistance development in gliomas. Should there be one in the future, more study of tumour dynamics is needed to translate pre-and-post therapy CSF ctDNA mutations in these markers into actionable clinical information.
What are the challenges to implementing liquid biopsy?
Sensitivity: The best reported sensitivity has mainly been in liquid biopsies of patients with advanced colon, ovarian and breast carcinomas, with sensitivity approaching 100% [32,65]. The fact that sensitivity increases in advanced disease is not surprising: the sensitivity of measuring ctDNA is reliant on tumour cell abundance, which in turn increases with overall tumour burden. CTCs, ctDNA and MVs have previously been detectable in different biofluids for a few tumour types, including adult infiltrating gliomas. While there has been success in detecting these biomarkers in the peripheral biofluids of GBM patients, and that their mutational profiles mirror those in the surgical specimen; the sensitivity of these liquid biopsies is lower for glioblastomas, with detection rates for ctDNA in GBM patients’ blood ranging from 10%–55% [15,29,31] and 20% to 77% for CTC’s in GBM patients [19,24]. While the sensitivity of peripheral biomarkers in glioblastoma does not approach that seen in colon, ovarian and breast carcinomas, this does not negate the role of utilising liquid biopsies in GBM patients, provided one understands the inherent limitations of our present liquid biopsy technology for GBM. It is also clear that there is a need to improve the technologies involved in regularly and reliably isolating and characterising these biomarkers. Larger studies in GBM investigating these peripherally detected biomarkers are warranted, in order to reliably determine their effects on clinical outcome.
Reliance on CSF for liquid biopsies: At present, the comparisons between CSF and plasma as an analytic fluid have demonstrated the clear superiority of the former, in part due to the blood-brain barrier inhibiting passage of biomarkers into the peripheral blood [30]. Lumbar puncture is relatively safe compared to a brain tumour biopsy, but it is still not a risk-free medical procedure. Until there is a sufficiently sensitive and methodologically validated method of extracting GBM biomarkers from peripheral blood, lumbar puncture may be considered an interim alternative method for studying these biomarkers.
Standardisation: Despite being a potentially powerful tool that enables non-invasive study of the tumour genome and proteome over time, major technological issues must be resolved before extraction of molecules from circulating fluid becomes a routine, validated test in glioblastoma. Various extraction, purification, quantification, and molecular sequencing methods have been reported in the literature, and these are far from being standardised and reproducible. Complicating matters further are the lack of consensus and validation studies on how to control pre-analytic variables such as timing, collection, and storage, given the short half-life of these molecules. Moreover, a patient’s comorbid or iatrogenic inflammatory conditions (sepsis, malnutrition and the like), which do fluctuate in cancer patients, can also introduce confounding variables that can affect the accuracy of the assay. Strategies for adjusting methodology and interpretation with respect to these confounding variables has not been described.
In addition, there is a need for standardisation of the preanalytic variables for liquid biopsy collection. For example, the analyte yield from different liquid biopsy isolation techniques can be influenced by freeze/thaw duration, elapsed time before processing, specimen haemolysis, preservative agent exposure, and storage conditions [37,66]. In the case of EVs, the method of vesicle counting can also yield different results [38], and this should be accounted for in downstream analytics. As the field of liquid biopsy is brain tumours is far from being validated, the lack of standard guidelines and therefore reproducibility of biomarker studies is a distinct problem. The variety of techniques used -microarrays, real-time quantitative polymerase chain reaction (qRT-PCR), RNAseq, and droplet digital PCR (ddPCR) – means each one carries its own advantages, limitations, and biases. Efforts should be placed in ensuring the continuity of technology used from discovery to full validation of the assay.
Technology development: In addition to the challenges from lack of standardisation and validation, the current workflow for biomarker analysis requires resource-intensive technologies, such as NGS platforms, BEAM-ing or ddPCR. These technologies have been validated for the study of excised tissue and allow us to explore a wide range of nucleic acids present in a sample with high sensitivity and reproducibility. The technical challenge in years to come will be to hone the sensitivity and reproducibility of these to a level that these can be deployed broadly into pathology laboratories, while at the same time keeping the cost low to make these widely available to patients.
There is a justified interest in liquid biopsy methods for monitoring glioblastoma, given its advantages over traditional tissue biopsies in its ability to capture tumoral heterogeneity and monitor changes in the brain tumour genome and proteome without repeated surgery, enabling better prognostication and monitoring of treatment response. A liquid biopsy can reveal tumour information before clinically apparent progression. However, the tumour morphology and its relation to the microenvironment still best studied in the tissue biopsy. Therefore, a liquid biopsy, rather than completely supplanting conventional tissue biopsy, aims to provide complementary information to improve the diagnosis and follow-up of GBM patients.
Studies have demonstrated CTCs, ctDNA and MVs as viable candidates for liquid biopsy analysis for a number of tumour types, and also have demonstrated that these biomarkers can be found in glioma patients, and that mutational profiles detected therein closely mirror that from the originating tumour. Important epigenetic changes such as MGMT promoter methylation can also be detected. There is an urgent need to improve the laboratory techniques used in reliably isolating and characterising these biomarkers. Hence, larger studies in GBM investigating these biomarkers and their retrieval techniques are warranted, with clinical follow-up to determine the effects on clinical outcome.
Currently, no clinically-validated circulating biomarkers for managing gliomas exist. One reason for the difficulty in identifying sufficiently reliable circulating biomarkers is because of the blood-brain barrier, which restricts the transportation of potentially diagnostically useful molecules from the neuraxis to the blood. Along with biological difficulties, there are technical limitations for the establishment of a role for circulating biomarkers in gliomas.
As an illustration of the difficulty of obtaining reproducible, reliable results, one may look at the detection rates obtained from CTC’s and ctDNA in glioma. In the few studies have been carried out by using brain- tumour-derived CTCs, and they show that the detection rates vary widely (from 20% to 77%) in GBM patients, depending on the isolation techniques used.
Similarly, the detection rates for ctDNA in GBM patients’ blood also vary widely (10%–55%).
However, technology is constantly improving and more recent studies are promising. For example, the first report detecting CTC clusters in GBM, by Krol et.al in 2018, indicates the capacity of GBM cell clusters to cross the BBB at levels sufficient to be detected [28].
GBM patients often develop resistance to treatment. Serial, minimally invasive blood or CSF sampling throughout treatment can help us detect specific tumour mutations and changes in DNA methylation pattern, which in turn might prove valuable for understanding a tumour’s biological response to treatment. These serial liquid biopsies can complement the current conventional methods used in managing GBM patients. Moreover, such serial sampling can help us monitor tumour progression and help distinguish it from pseudo progression. When different biofluid sources are compared—for example, ctDNA detected in blood or in CSF—CSF appears to show a higher yield, possibly owing to the proximity of CSF to the brain. Nevertheless, CSF collection is still invasive and not without risk, compared with blood collection.
Extracellular vesicle detection in GBM is showing promise at delivering clinically significant prognostic data by studying transcriptional products, at least where large gene deletions are involved, such as the EGFRvIII deletion variant in tumour tissue. [44]. There is also potential for extracellular vesicles to be a valuable method of studying transcriptional products such as miRNA-21, and mi-RNA 181 [46,49,50].
The concept of detecting CTCs, ctDNA and exosomes that carry predictive markers for GBM, such as IDH1, MGMT and EGFRvIII, is interesting and we have provided proof-of-principle for the non-invasive detection of the more well-known IDH1R132H mutation, as well as a new, non-canonical IDH-2 mutation of unknown significance emerging after adjuvant therapy. Going forward, the extant literature indicates the feasibility of detecting more of these molecules in body fluids as we do for normal surgical biopsies. Due to each of the detected biomarkers having their own advantages and limiations (summarised in table 2), a panel tests to look for each of them might be beneficial. The rapid advances in liquid biopsy applications have spurred the investigation of a number of different and complementary biomarkers, which might better inform us on the tumour status and provide useful, complementary information to guide treatment when the quality and quantity of treated tumour material is less than optimal. The potential for using this method to easily divide patients into molecular subgroups for target therapies, and provide data on therapeutic efficacy over time, is exciting. More studies are needed to further develop the reliability of such assays and ultimately to advance future clinical applications as well as to bring down costs to an affordable level for wide application.
| Table 2: Summary of advantages and limitations of various analytes in liquid biopsy specimens | |||
| LIQUID BIOPSY ANALYTE | Advantages | Limitations | Ref |
| CTC's |
|
|
(15–17,20,69) |
| ctDNA |
|
|
(15,29,41,42) |
| EV |
|
|
(22,28,34,44,45,70) |
- Hartmann C, Hentschel B, Tatagiba M, Schramm J, Schnell O, et al. Molecular Markers in Low-Grade Gliomas: Predictive or Prognostic? Clin Cancer Res. 2011; 17: 4588–4599. Pubmed: http://www.ncbi.nlm.nih.gov/pubmed/21558404
- Reuss DE, Mamatjan Y, Schrimpf D, Capper D, Hovestadt V, et al. IDH mutant diffuse and anaplastic astrocytomas have similar age at presentation and little difference in survival: a grading problem for WHO. Acta Neuropathol. 2015; 129: 867–873. PubMed: https://pubmed.ncbi.nlm.nih.gov/25962792/
- Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016; 131: 803–820. PubMed: https://pubmed.ncbi.nlm.nih.gov/27157931/
- Sahm F, Schrimpf D, Jones DTW, Meyer J, Kratz A, et al. Next-generation sequencing in routine brain tumor diagnostics enables an integrated diagnosis and identifies actionable targets. Acta Neuropathol. 2016; 131: 903–910. PubMed: https://pubmed.ncbi.nlm.nih.gov/26671409
- Li KW, Ng HK. How does one do next-generation sequencing for brain tumors in the clinical laboratories? Glioma. 2018; 1: 149.
- Kim H, Zheng S, Amini SS, Virk SM, Mikkelsen T, et al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res. 2015; 25: 316–227. PubMed: https://pubmed.ncbi.nlm.nih.gov/25650244/
- Hartmann C, Hentschel B, Simon M, Westphal M, Schackert G, et al. Long-term survival in primary glioblastoma with versus without isocitrate dehydrogenase mutations. Clin Cancer Res. 2013; 19: 5146–5157. PubMed: https://pubmed.ncbi.nlm.nih.gov/23918605/
- Sanson M, Marie Y, Paris S, Idbaih A, Laffaire J, et al. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol. 2009; 27: 4150–4154. PubMed: https://pubmed.ncbi.nlm.nih.gov/19636000/
- Yan H, Williams D, Jin G, Mclendon R, Rasheed BA, et al. IDH1 and IDH2 Mutations in Gliomas. N Engl J Med. 2009; 360: 765–773. PubMed: https://pubmed.ncbi.nlm.nih.gov/19228619/
- Loo HK, Mathen P, Lee J, Camphausen K. Circulating biomarkers for high-grade glioma. Biomark Med. 2019; 13: 161–165. PubMed: https://pubmed.ncbi.nlm.nih.gov/30806515/
- Brat DJ, Verhaak RGW, Aldape KD, Yung WKA, Salama SR, et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. 2015; 372: 2481–2498. PubMed: http://www.nejm.org/doi/10.1056/NEJMoa1402121
- Wang SR, Malik S, Tan IB, Chan YS, Hoi Q, et al. Technical Validation of a Next-Generation Sequencing Assay for Detecting Actionable Mutations in Patients with Gastrointestinal Cancer. J Mol Diagnostics. 2016; 18: 416–424. PubMed: https://pubmed.ncbi.nlm.nih.gov/26970585/
- Horbinski C. What do we know about IDH1/2 mutations so far, and how do we use it? Acta Neuropathol. 2013; 125: 621–636. PubMed: https://pubmed.ncbi.nlm.nih.gov/23512379/
- Kruser TJ, Mehta MP, Robins HI. Pseudoprogression after glioma therapy: A comprehensive review. Expert Rev Neurother. 2013; 13: 389–403. PubMed: https://pubmed.ncbi.nlm.nih.gov/23545054/
- Westphal M, Lamszus K. Circulating biomarkers for gliomas. Nat Rev Neurol. 2015;11: 556–566. PubMed: https://pubmed.ncbi.nlm.nih.gov/26369507/
- Cabel L, Proudhon C, Gortais H, Loirat D, Coussy F, et al. Circulating tumor cells: clinical validity and utility. Int J Clin Oncol. 2017; 22: 421–430. PubMed: https://pubmed.ncbi.nlm.nih.gov/28238187/
- Choon AW T. Clinical Utility of Circulating Tumor Cells – A Clinician’s Current View. Hematol Med Oncol. 2017; 2: 1–9.
- Sottoriva A, Spiteri I, Piccirillo SGM, Touloumis A, Collins VP, et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci. 2013; 110: 4009–1014. PubMed: https://www.pnas.org/content/110/10/4009
- MacArthur KM, Kao GD, Chandrasekaran S, Alonso-Basanta M, Chapman C, et al. Detection of brain tumor cells in the peripheral blood by a telomerase promoter-based assay. Cancer Res. 2014; 74: 2152–2159. PubMed: https://pubmed.ncbi.nlm.nih.gov/24525740/
- Kros JM, Mustafa DM, Dekker LJM, Smitt PAES, Luider TM, et al. Circulating glioma biomarkers. Neuro Oncol. 2015; 17: 343–360. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4483097/
- Saenz-Antoñanzas A, Auzmendi-Iriarte J, Carrasco-Garcia E, Moreno-Cugnon L, Ruiz I, et al. Liquid biopsy in glioblastoma: Opportunities, applications and challenges. Cancers (Basel). 2019; 11: 950. PubMed: https://pubmed.ncbi.nlm.nih.gov/31284524/
- Müller Bark J, Kulasinghe A, Chua B, Day BW, Punyadeera C. Circulating biomarkers in patients with glioblastoma. Br J Cancer. 2020; 122: 295-305.. PubMed: https://pubmed.ncbi.nlm.nih.gov/31666668/
- Orlic L, Sladoje-Martinovic B, Mikolasevic I, Zupan Z, Racki S. Patients with primary brain tumors as organ donors. BANTAO J. 2015; 13: 34–38.
- Müller C, Holtschmidt J, Auer M, Heitzer E, Lamszus K, et al. Cancer: Hematogenous dissemination of glioblastoma multiforme. Sci Transl Med. 2014; 6: 247ra101. PubMed: https://pubmed.ncbi.nlm.nih.gov/25080476/
- Jackson D. Rapp HM, Schneiderhan TM, Michael Sabel Anne Hayman Axel SchererPatric Kröpil , Wilfried Budach , Usha Kretschmar , Peter Arne Gerber Sujit Prabhu , Lawrence E. Ginsberg Edwin Bölke CM. Glioblastoma Multiforme Metastasis Outside the CNS: Three Case Reports and Possible Mechanisms of Escape Introduction. J Clin Oncol. 2012; 29: 2011–2013.
- Ferreira MM, Ramani VC, Jeffrey SS. Circulating tumor cell technologies. Mol Oncol. 2016; 10: 374–394. www.nature.com/npjprecisiononcology%0Ahttp://www.nature.com/articles/s41698-017-0028-8
- Lombard A, Goffart N, Rogister B. Glioblastoma circulating cells: Reality, trap or illusion? Stem Cells Int. 2015; 2015. PubMed: https://pubmed.ncbi.nlm.nih.gov/26078762/
- Krol I, Castro-Giner F, Maurer M, Gkountela S, Szczerba BM, et al. Detection of circulating tumour cell clusters in human glioblastoma. Br J Cancer. 2018; 119: 487–491. PubMed: https://pubmed.ncbi.nlm.nih.gov/30065256/
- Piccioni DE, Lanman RB, Nagy RJ, Talasaz A, Pingle SC, et al. Analysis of cell-free circulating tumor DNA in patients with glioblastoma and other primary brain tumors. J Clin Oncol. 2015; 33: 11072–11072. https://ascopubs.org/doi/10.1200/jco.2015.33.15_suppl.11072
- Wang Y, Springer S, Zhang M, McMahon KW, Kinde I, et al. Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc Natl Acad Sci. 2015; 112: 9704–9709. PubMed: https://pubmed.ncbi.nlm.nih.gov/26195750/
- Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, et al. Detection of Circulating Tumor DNA in Early- and Late-Stage Human Malignancies. Sci Transl Med. 2014; 62 24ra24-224ra24. http://stm.sciencemag.org/cgi/doi/10.1126/scitranslmed.3007094
- Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008; 14: 985–990. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2820391/ b
- Diaz LA, Bardelli A. Liquid biopsies: Genotyping circulating tumor DNA. J Clin Oncol. 2014; 32: 579–586. PubMed: https://pubmed.ncbi.nlm.nih.gov/24449238/
- Chen WW, Balaj L, Liau LM, Samuels ML, Kotsopoulos SK, et al. Beaming and droplet digital pcr analysis of mutant idh1 mrna in glioma patient serum and cerebrospinal fluid extracellular vesicles. Mol Ther - Nucleic Acids. 2013; 2: e109. PubMed: https://pubmed.ncbi.nlm.nih.gov/23881452/
- Ghosh RK, Pandey T, Dey P. Liquid biopsy: A new avenue in pathology. Cytopathology. 2019; 30: 138–143. PubMed: https://pubmed.ncbi.nlm.nih.gov/30485558/
- Bertero L, Siravegna G, Rudà R, Soffietti R, Bardelli A, et al. Review: Peering through a keyhole: liquid biopsy in primary and metastatic central nervous system tumours. Neuropathol Appl Neurobiol. 2019; 45: 655–670. PubMed: https://pubmed.ncbi.nlm.nih.gov/30977933
- Schwaederle M, Chattopadhyay R, Kato S, Fanta PT, Kimberly C, et al. Genomic alterations in circulating tumor DNA from diverse cancer patients identified by next-generation sequencing. Cancer Res. 2017; 77: 5419–5427. PubMed: https://pubmed.ncbi.nlm.nih.gov/28807936/
- Piccioni DE, Achrol AS, Kiedrowski LA, Banks KC, Boucher N, et al. Analysis of cell-free circulating tumor DNA in 419 patients with glioblastoma and other primary brain tumors. CNS Oncol. 2019; 8: CNS34. PubMed: https://pubmed.ncbi.nlm.nih.gov/30855176/
- Brennan CW, Verhaak RGW, McKenna A, Campos B, Noushmehr H, et al. The Somatic Genomic Landscape of Glioblastoma. Cell. 2013; 155: 462–477. https://linkinghub.elsevier.com/retrieve/pii/S0092867413012087
- WANG Z, JIANG W, WANG Y, GUO Y, CONG Z, et al. MGMT promoter methylation in serum and cerebrospinal fluid as a tumor-specific biomarker of glioma. Biomed Reports. 2015; 3: 543–548. PubMed: https://pubmed.ncbi.nlm.nih.gov/26171163/
- De Mattos-Arruda L, Mayor R, Ng CKY, Weigelt B, Martínez-Ricarte F, et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat Commun. 2015; 6: 8839. PubMed: https://pubmed.ncbi.nlm.nih.gov/26554728/
- Miller AM, Shah RH, Pentsova EI, Pourmaleki M, Briggs S, et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature. 2019; 565: 654–658. PubMed: https://pubmed.ncbi.nlm.nih.gov/30675060/
- Li I, Nabet BY. Exosomes in the tumor microenvironment as mediators of cancer therapy resistance. Mol Cancer. 2019; 18: 32. PubMed: https://pubmed.ncbi.nlm.nih.gov/30823926/
- Skog J, Würdinger T, van Rijn S, Meijer DH, Gainche L, et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008; 10: 1470–1476. PubMed: https://pubmed.ncbi.nlm.nih.gov/19011622/
- Shao H, Chung J, Balaj L, Charest A, Bigner DD, et al. Protein typing of circulating microvesicles allows real-time monitoring of glioblastoma therapy. Nat Med. 2012; 18: 1835–1840. http://www.nature.com/articles/nm.2994
- Banelli B, Forlani A, Allemanni G, Morabito A, Pistillo MP, et al. MicroRNA in glioblastoma: An overview. Int J Genomics. 2017; 2017: 7639084. PubMed: https://pubmed.ncbi.nlm.nih.gov/29234674/
- Bader AG, Brown D, Winkler M. The Promise of MicroRNA Replacement Therapy. Cancer Res. 2010; 70: 7027–7030. PubMed: https://pubmed.ncbi.nlm.nih.gov/20807816/
- Li CCY, Eaton SA, Young PE, Lee M, Shuttleworth R, et al. Glioma microvesicles carry selectively packaged coding and noncoding RNAs which alter gene expression in recipient cells. RNA Biol. 2013; 10: 1333–1344. PubMed: https://pubmed.ncbi.nlm.nih.gov/23807490/
- Shi R, Wang PY, Li XY, Chen JX, Li Y, et al. Exosomal levels of miRNA-21 from cerebrospinal fluids associated with poor prognosis and tumor recurrence of glioma patients. Oncotarget. 2015; 6: 26971–26981. PubMed: https://pubmed.ncbi.nlm.nih.gov/26284486/
- Qu S, Guan J, Liu Y. Identification of microRNAs as novel biomarkers for glioma detection: A meta-analysis based on 11 articles. J Neurol Sci. 2015; 348: 181–187. PubMed: https://pubmed.ncbi.nlm.nih.gov/25510379/
- Zhang W, Zhang J, Hoadley K, Kushwaha D, Ramakrishnan V, et al. MiR-181d: Predictive glioblastoma biomarker that downregulates MGMT expression. Neuro Oncol. 2012; 14: 712–719. PubMed: https://pubmed.ncbi.nlm.nih.gov/22570426/
- Verbeek B, Southgate TD, Gilham DE, Margison GP. O6-Methylguanine-DNA methyltransferase inactivation and chemotherapy. Br Med Bull. 2008; 85: 17–33. PubMed: https://pubmed.ncbi.nlm.nih.gov/18245773/
- Tang K, Gardner S, Snuderl M. The Role of Liquid Biopsies in Pediatric Brain Tumors. J Neuropathol Exp Neurol. 2020; 79: 934-940. PubMed: https://pubmed.ncbi.nlm.nih.gov/32766689/
- Tuxen MK, Sölétormos G, Dombernowsky P. Serum tumour marker CA 125 in monitoring of ovarian cancer during first-line chemotherapy. Br J Cancer. 2001; 84: 1301–1307. PubMed: https://pubmed.ncbi.nlm.nih.gov/11355938/
- Ito K, Hibi K, Ando H, Hidemura K, Yamazaki T, et al. Usefulness of analytical CEA doubling time and half-life time for overlooked synchronous metastases in colorectal carcinoma. Jpn J Clin Oncol. 2002; 32: 54–58. PubMed: https://pubmed.ncbi.nlm.nih.gov/11948229/
- Shinozaki M, O’Day SJ, Kitago M, Amersi F, Kuo C, et al. Utility of circulating B-RAF DNA mutation in serum for monitoring melanoma patients receiving biochemotherapy. Clin Cancer Res. 2007; 13: 2068–2074. PubMed: https://pubmed.ncbi.nlm.nih.gov/17404088/
- Dawson SJ, Tsui DWY, Murtaza M, Biggs H, Rueda OM, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013; 368: 1199–1209. PubMed: https://pubmed.ncbi.nlm.nih.gov/23484797/
- Thomsen CEB, Appelt AL, Andersen RF, Lindebjerg J, Jensen LH, et al. The prognostic value of simultaneous tumor and serum RAS/RAF mutations in localized colon cancer. Cancer Med. 2017; 6: 928–936. PubMed: https://pubmed.ncbi.nlm.nih.gov/28378527/
- Rudà R, Reifenberger G, Frappaz D, Pfister SM, Laprie A, et al. EANO guidelines for the diagnosis and treatment of ependymal tumors. Neuro Oncol. 2018; 20: 445–456. PubMed: https://pubmed.ncbi.nlm.nih.gov/29194500/
- Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell. 2017; 168: 707–723. PubMed: https://pubmed.ncbi.nlm.nih.gov/28187290/
- Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012; 486: 532–536. PubMed: https://pubmed.ncbi.nlm.nih.gov/22722830/
- Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: Learning from lung cancer. Nat Rev Clin Oncol. 2014; 11: 473–481. PubMed: https://pubmed.ncbi.nlm.nih.gov/24981256/
- Serrano C, George S, Valverde C, Olivares D, García-Valverde A, et al. Novel Insights into the Treatment of Imatinib-Resistant Gastrointestinal Stromal Tumors. Target Oncol. 2017; 12: 277–288. PubMed: https://pubmed.ncbi.nlm.nih.gov/28478525/
- Murtaza M, Dawson SJ, Tsui DWY, Gale D, Forshew T, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013; 497: 108–112. PubMed: https://pubmed.ncbi.nlm.nih.gov/23563269/
- Higgins MJ, Jelovac D, Barnathan E, Blair B, Slater S, et al. Detection of tumor PIK3CA status in metastatic breast cancer using peripheral blood. Clin Cancer Res. 2012; 18: 3462–3469. PubMed: https://pubmed.ncbi.nlm.nih.gov/22421194/
- Akers JC, Ramakrishnan V, Yang I, Hua W, Mao Y, et al. Optimizing preservation of extracellular vesicular miRNAs derived from clinical cerebrospinal fluid. Cancer Biomarkers. 2016; 17: 125–132. PubMed: https://pubmed.ncbi.nlm.nih.gov/27062568/
- Fritz JV, Heintz-Buschart A, Ghosal A, Wampach L, Etheridge A, et al. Sources and Functions of Extracellular Small RNAs in Human Circulation. Annu Rev Nutr. 2016; 36: 301–336. PubMed: https://pubmed.ncbi.nlm.nih.gov/27215587/
- Akers JC, Ramakrishnan V, Nolan JP, Duggan E, Fu CC, et al. Comparative analysis of technologies for quantifying extracellular vesicles (EVs) in clinical cerebrospinal fluids (CSF). PLoS One. 2016; 11: e0149866. PubMed: https://pubmed.ncbi.nlm.nih.gov/26901428/
- Gao F, Cui Y, Jiang H, Sui D, Wang Y, et al. Circulating tumor cell is a common property of brain glioma and promotes the monitoring system. Oncotarget. 2016; 7: 71330–71340. PubMed: https://pubmed.ncbi.nlm.nih.gov/27517490/
- Kros JM, Huizer K, Hernández-Laín A, Marucci G, Michotte A, et al. Evidence-based diagnostic algorithm for glioma: Analysis of the results of pathology panel review and molecular parameters of EORTC 26951 and 26882 trials. J Clin Oncol. 2015; 33: 1943–1950. PubMed: https://pubmed.ncbi.nlm.nih.gov/25918297/